
Pathogenetic Mechanisms Underlying the Development and Progression of Interstitial Lung Diseases
Interstitial lung processes (ILP) represent an umbrella term for a wide range of nosological units that differ from each other in clinical course, prognosis, and pathogenesis. Most ILPs are characterized by inflammation or fibrosis of the interstitial space, leading to impaired gas exchange at the alveolocapillary membrane.
Diverse Range of ILP
Interstitial lung processes include more than 200 individual diseases, most of which (though not all) are associated with primary damage to the lung interstitium. ILPs also include diseases linked to primary damage to the alveoli. An example is pulmonary alveolar proteinosis, characterized by excessive accumulation of surfactant components in the alveoli and distal airways with minimal inflammatory response and interstitial fibrosis. However, most ILPs are associated with inflammation or fibrosis of the interstitium or a combination of both pathogenetic mechanisms, with primary inflammation progressing to pulmonary fibrosis.
Overview of Pathogenetic Mechanisms
Inflammatory processes within interstitial lung processes can be caused by various reasons − the most common being autoimmune diseases. Among the diseases with the best-understood pathogenesis is rheumatoid arthritis, which in genetically susceptible individuals is associated with the formation of antibodies against abnormally citrullinated structural proteins. The presence of these antibodies leads to the activation of specialized macrophages and stromal cells and the development of an inflammatory response leading to the production of various cytokines (e.g., IL-6, TNF-α). However, it is still unclear whether the interstitial damage is caused by the direct effect of antibodies or by the paracrine effect of circulating cytokines and growth factors.
Autoantibodies and Interstitial Damage
Similar pathogenetic features with different autoantibody repertoires are shared by other autoimmune diseases, including systemic scleroderma or idiopathic inflammatory myopathy. Interestingly, the likelihood of developing pulmonary disease and its type (such as organizing pneumonia, usual interstitial pneumonia, nonspecific interstitial pneumonia) in these units is determined by the presence of specific autoantibodies, suggesting a significant role of direct interstitial damage by autoantibodies.
Granuloma Formation
Another common manifestation of inflammatory processes is the formation of granulomas composed of aggregated macrophages and other immune system cells. Granulomas without caseous necrosis are a dominant characteristic of the multisystem disease − sarcoidosis, which affects the lungs in more than 90% of individuals. Granuloma formation is also characteristic of hypersensitivity pneumonitis, which occurs after repeated exposure to antigens from the external environment (most commonly bird and mold proteins).
How Does Pulmonary Fibrosis Develop?
The pathogenesis of interstitial fibrosis is best understood in the context of idiopathic pulmonary fibrosis. The development of fibrotic changes likely depends on a triad of factors:
- lifelong excessive epithelial damage due to exposure to inhaled harmful substances
- aging
- genetic susceptibility
The combination of these factors leads to premature aging of alveolar epithelial stem cells and aberrant healing of epithelial injury. The aging of stem cells results in the failure of proliferation and repopulation of the alveolar epithelium, followed by denudation of the basement membrane. This disrupts the cascade of healing processes, leading to a disbalance of profibrotic and antifibrotic growth factors. Profibrotic growth factors activate several types of inflammatory cells, such as macrophages, fibroblasts, epithelial cells, and endothelial cells.
The result of their activation is the excessive production of collagen and extracellular matrix. Progressive alveolar damage worsens conditions for gas exchange and causes aberrant remodeling of pulmonary vessels, with the subsequent risk of developing secondary pulmonary hypertension. Changes in the composition of the extracellular matrix and increasing stiffness of lung tissue lead to further progression of fibrosis, indicating that pulmonary fibrosis may self-sustain from a certain level of severity regardless of the initial trigger.
In other diseases, the pathogenesis of fibrosis is less explored; the initial trigger may be different, but the pathway leading to fibrosis may overlap and be identical to the pathogenesis of idiopathic pulmonary fibrosis.
Figure Pathogenesis of Interstitial Lung Diseases
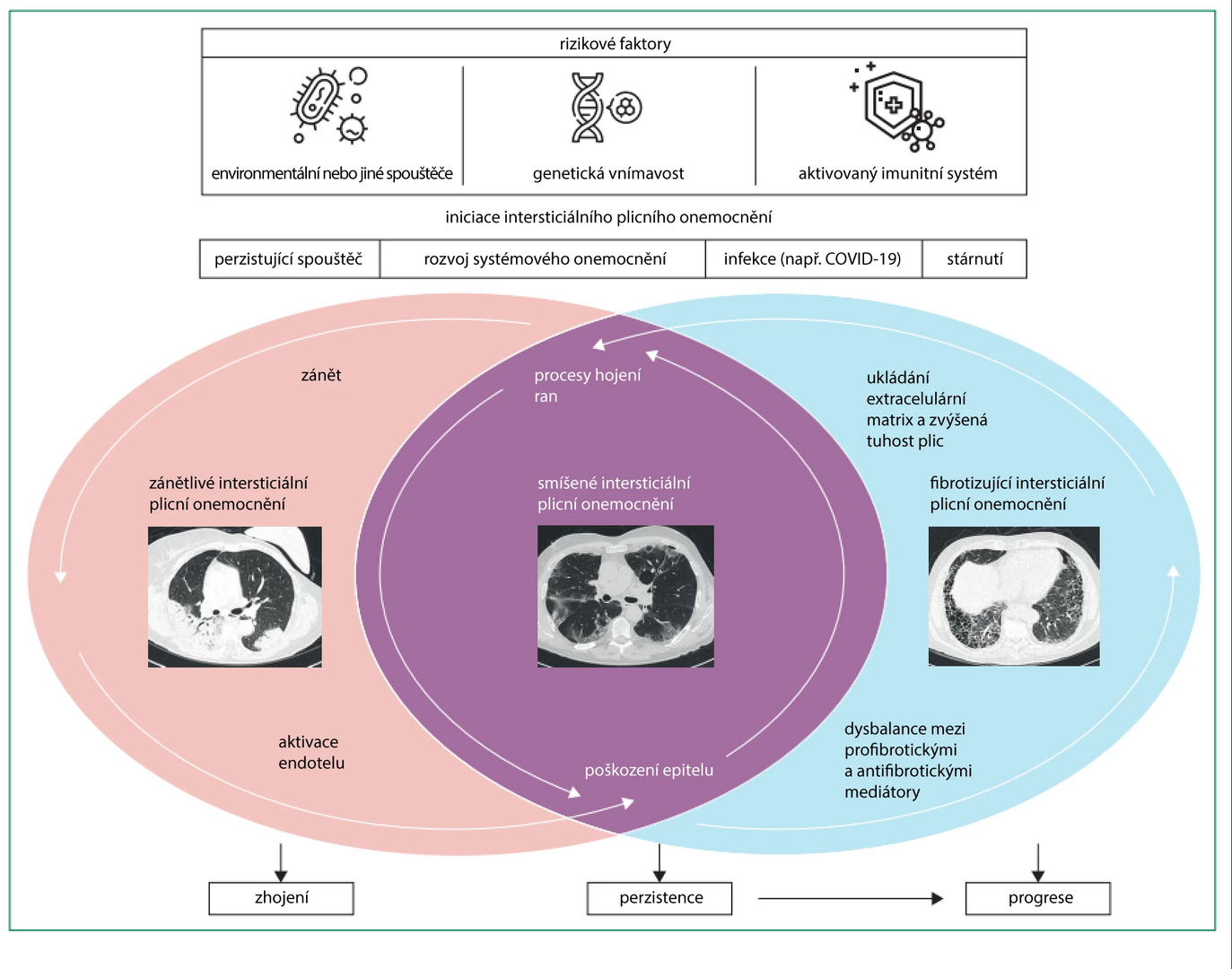
Other Mechanisms of ILP Development
Another pathogenetic group is the so-called orphan ILP, which exhibit distinct disease phenotypes. An example is the rare disease lymphangioleiomyomatosis, which is caused by the constitutive activation of mTOR signaling in smooth muscle cells.
The aforementioned pulmonary alveolar proteinosis is caused by the accumulation of surfactant components and macrophages due to a failure in the GM-CSF signaling pathway.
Langerhans cell histiocytosis is caused by the proliferation of a subset of dendritic cells associated with somatic mutations in the BRAF and MAPK genes.
Conclusion
Interstitial lung processes represent a heterogeneous group of nosological units usually associated with inflammatory processes or fibrosis, or a combination of inflammation and fibrosis. The identification of pathogenetic mechanisms has enabled significant advancements in therapy, such as the administration of antifibrotic drugs, which significantly improve the prognosis of patients.
(holi)
Source: Wijsenbeek M., Suzuki A., Maher T. M. Interstitial lung diseases. Lancet 2022; 400 (10354): 769–786, doi: 10.1016/S0140-6736(22)01052-2.
Did you like this article? Would you like to comment on it? Write to us. We are interested in your opinion. We will not publish it, but we will gladly answer you.