
Treatment for Transthyretin Cardiac Amyloidosis is Now More Accessible to Czech Patients
Transthyretin cardiac amyloidosis is a serious progressive disease that was until recently considered untreatable. In recent years, however, new treatment options targeting the underlying pathology of the disease have emerged, which, along with advances in non-invasive diagnostics, offer patients hope for a longer and better-quality life.
Cardiac Amyloidosis
Transthyretin cardiac amyloidosis (ATTR-CM) is a fatal disease caused by the deposition of amyloid fibrils formed by misfolded transthyretin protein transthyretin (TTR). Misfolding of TTR can be genetically driven, due to mutations in the TTR gene; in older individuals with non-mutated (wt - wild-type) TTR gene, it is associated with aging.1 The median survival from the time of diagnosis for wtATTR patients is about 3.5 years.2
Targeting the Molecular Basis
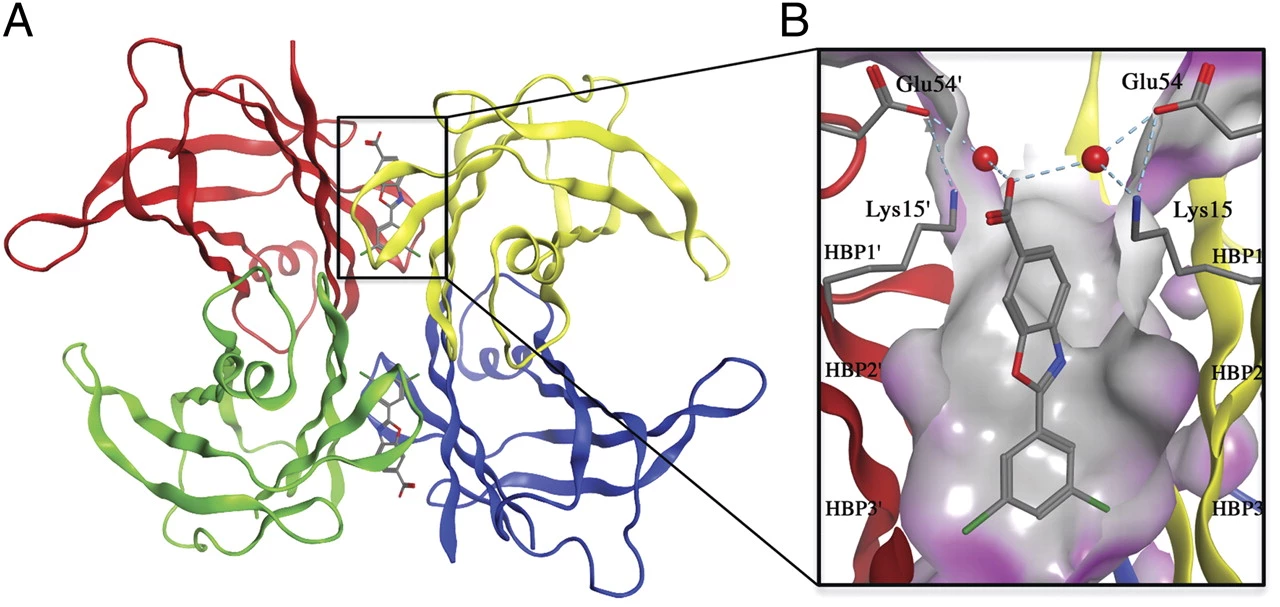
ATTRwt stems from TTR dissociation from the native tetramer, resulting monomers then aggregate into amyloid fibrils, which deposit in various tissues. Hence, therapy now utilizes tafamidis - a selective stabilizer that prevents tetramer dissociation and subsequent amyloidogenesis (see fig.).
Fig. Crystal structure of tafamidis bound to TTR tetramer3:
A) Ribbon diagram showing individual TTR monomers in different colors.
B) Detail of the tafamidis molecule in the hydrophobic binding cavity.
The efficacy of tafamidis was demonstrated in a double-blind, placebo-controlled phase III clinical study ATTR-ACT (Tafamidis in Transthyretin Cardiomyopathy Clinical Trial), where patients using this therapy over 30 months showed a significant reduction in mortality (hazard ratio [HR] 0.70; 95% confidence interval [CI] 0.51–0.96) and cardiovascular-related hospitalizations (relative risk [RR] 0.68; 95% CI 0.56–0.81) compared to the placebo group.4
Starting Treatment Early is Crucial
The latest analysis of data from the open-label extension of this study showed significantly better survival for patients who took tafamidis continuously from study entry (median follow-up 58.5 months; 44.9% of patients in this arm died) compared to those who started treatment after the 30-month pivotal study period during which they received placebo (median follow-up 57.1 months; 62.7% deaths). The continuous treatment group experienced a 41% reduction in the risk of all-cause mortality. Even patients who started treatment later displayed improved overall survival compared to the extrapolated model curve for ongoing placebo use.1
Reimbursement by Health Insurance
The data from the above-mentioned studies underscore how crucial early diagnosis and prompt treatment initiation are for patient survival. As of June 1, 2023, tafamidis is reimbursed from public health insurance funds for adult patients with wtATTR-CM. Treatment must be initiated while the patient is in NYHA functional class I-II. Therapy must be discontinued if the patient experiences a sustained deterioration to NYHA class IV, a drop in GFR to < 25 ml/min/1.73 m2, progressive severe hepatopathy, uncontrollable severe malnutrition, severe dementia, debilitating neurological disease, or other serious illnesses with a life expectancy of less than 12 months.5
Where to Refer the Patient?
Reimbursed treatment is available within the network of centers for the diagnosis and treatment of cardiac amyloidosis.6 Currently, there are four such centers:
- Cardiology Clinic IKEM Prague
- 2nd Department of Internal Medicine – Department of Cardiology and Angiology, 1st Faculty of Medicine, Charles University and General University Hospital in Prague
- 1st Internal Cardioangiologic Clinic, Faculty of Medicine MU and St. Anne University Hospital in Brno
- Department of Internal Medicine I – Cardiology, Faculty of Medicine UP and University Hospital Olomouc
(este)
Sources:
1. Elliott P., Drachman B. M., Gottlieb S. S. et al. Long-term survival with tafamidis in patients with transthyretin amyloid cardiomyopathy. Circ Heart Fail 2022; 15 (1): e008193, doi: 10.1161/CIRCHEARTFAILURE.120.008193.
2. Ruberg F. L., Maurer M. S., Judge D. P. et al. Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: the Transthyretin Amyloidosis Cardiac Study (TRACS). Am Heart J 2012; 164 (2): 222–228.e1, doi: 10.1016/j.ahj.2012.04.015.
3. Bulawa C. E., Connelly S., Devit M. et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci USA 2012; 109 (24): 9629–9634, doi: 10.1073/pnas.1121005109.
4. Maurer M. S., Schwartz J. H., Gundapaneni B. et al.; ATTR-ACT Study Investigators. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 2018; 379 (11): 1007–1016, doi: 10.1056/NEJMoa1805689.
5. Decision on the determination of the maximum price and the amount and conditions of reimbursement from public health insurance for the medicinal product intended for the treatment of rare diseases Vyndaqel. SÚKL, 31 March 2023. Available at: www.sukl.eu/modules/procedures/detail.php?spzn=SUKLS117667%2F2022
6. Treatment options for cardiac amyloidosis. Pharmacotherapeutic Information 2021; 3: 1–4. Available at: www.sukl.cz/file/95387_1_1
7. SPC Vyndaqel. Available at: www.ema.europa.eu/en/documents/product-information/vyndaqel-epar-product-information_cs.pdf
Did you like this article? Would you like to comment on it? Write to us. We are interested in your opinion. We will not publish it, but we will gladly answer you.